云南C57小鼠原代细胞分离培养优势
上期我们主要讲解了细胞凋亡的早期检测方法,这期主要讲解一下细胞凋亡晚期中,核酸内切酶(某些Caspase的底物)在核小体之间剪切核DNA,产生大量长度在180-200bp的DNA的片段。1、凋亡DNALadder检测试剂盒细胞经处理后,采用常规方法分离提纯DNA,进行琼脂糖凝胶和溴化乙啶染色,在凋亡细胞群中可观察到典型的DNAladder。QuickApoptoticDNALadderDetectionKit可以快速(约1-2小时)、灵敏地检测凋亡细胞中的DNA的片段。2、TUNEL-basedDNA的片段分析试剂盒细胞凋亡中,染色体DNA双链断裂或单链断裂而产生大量的粘性3-OH末端,可在脱氧核糖核苷酸末端转移酶(TdT)的作用下,将脱氧核糖核苷酸和荧光素、过氧化物酶、碱性磷酸酶或生物素形成的衍生物标记到DNA的3-末端,从而可进行凋亡细胞的检测。Apo-BrdUTM分析使用2种颜色的TUNEL方法用流式细胞仪检测凋亡细胞中DNA条带。采用的BrdU比生物素标记的或地高辛(digoxigenylated)标记的方法灵敏度更高。3、Caspase分析试剂和试剂盒Caspase在细胞凋亡中发挥着重要的作用,属于天冬氨酸蛋白酶。其中caspase-3为关键的执行分子,它在凋亡信号传导的许多途径中发挥功能。在正常状态下,caspase家族都以无活性的酶原。肝星状细胞参与了肝脏的纤维化、炎症发生和免疫紊乱等大多数病理生理过程。云南C57小鼠原代细胞分离培养优势
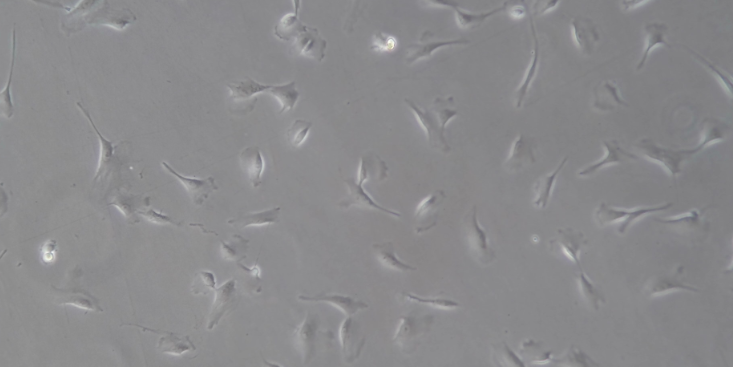
并进质粒抽提。GAG质粒和VSV质粒同样可以转化至感受态细菌DH5α中,并进行无内质粒抽提。质粒抽提后冷冻于-20℃保存。2、慢病毒包装1)使用DMEM完全培养基培养6cm皿HEK293T至汇合度为70~80%。采用opti-MEM和Lipo3000分别转染含有目的基因的pMSCV-eGFP、VSV、GAG质粒及对照载体,每皿加入脂质体-质粒转染混悬液按购买脂质体相关说明书操作定量。继续培养24h。2)24小时后,将培养基更换为新鲜的DMEM完全培养基,放进细胞培养箱继续培养48~72h。3)48~72h后收集上层培养液,并过μm滤膜,采用ELISA法对所获得的慢病毒载体进行病毒滴度测定。如不及时使用可以冻存于-80℃。3、慢病毒转染1)转染前1天将细胞接种6孔培养板,时细胞的融合率约为50%,前需换液,加入1mLDMEM完全培养基。2)病毒冰浴融化后加入相应体积的病毒液及聚凝胺(Polybrene),混匀后放入37℃孵箱中继续培养3)4h后补充1mL培养基,14h后换液(24h内换液即可)。4)病毒72h后用倒置显微镜观察荧光,监测效率,出现较多荧光时将等量的转染细胞和未转染细胞分别加入等浓度Puromycin(Puromycin或其他筛选浓度需要事先摸索)。5)待未转染细胞全部死亡并且可观察到满意荧光量时,降低Puromycin浓度培养。青海为什么原代细胞分离培养哪家好大鼠主动脉内皮细胞(aortic endothelial cells)组成了主动脉内壁,并持续受到血流剪切应力的影响。
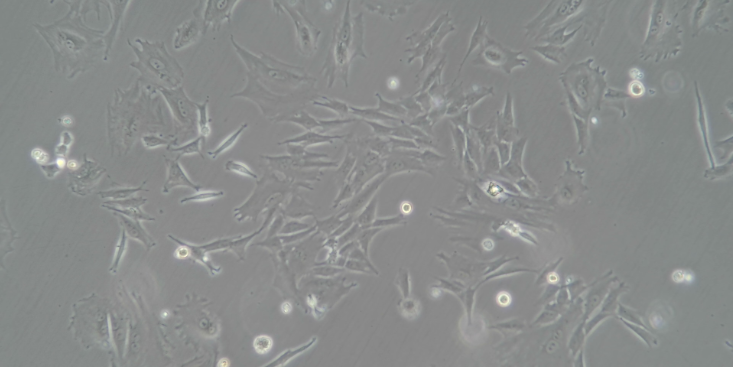
含血清完全培养液在2-8℃保存,需在1周内用完;5.增加接种细胞起始浓度;6.换用新的保种细胞;7.分离培养物,检测支原体。清洁支架和培养箱。如发现支原体污染,丢弃培养物。培养细胞生长不好可能原因细胞本身的状态1.细胞传代次数多,细胞老化;2.细胞的接种量:接种量过低,细胞生长缓慢;3.细胞传代时间过晚:细胞中毒,影响传代后的细胞生长;4.胰酶消化时间过长或过短:时间过长,细胞死亡;时间过短,细胞未完全分离而成团,细胞死亡;5.细胞的冻存与复苏:慢冻速溶。污染1.支原体污染;2.霉菌污染。培养基或血清1.更换血清或培养基之前未进行验证;2.选择的培养基是否合适;3.培养基配制是否合适;4.培养基配制是否准确无误。培养环境;2.培养箱或摇床温度控制是否正确。解决方法根据以上四个方面的可能原因,做出针对性的解决方案1.注意细胞的本身状态:如传代次数,接种量等;2.避免产生污染(用正规,合法,可朔源的血清);3.要用合适的血清或培养基,经过验证;4.注意实验室的环境。培养细胞死亡可能原因1.培养箱内无CO2;2.培养箱内温度波动太大;3.细胞冻存或复苏过程中损伤;4.培养液渗透压不正确;5.培养液中有毒代谢产物堆积。解决方法1.检测培养箱内CO2。
具体选用何种培养器皿取决于细胞生长密度需求(启动正常生长所需的密度)。二.细胞换液培养1、全量换液:把所有的旧培养液移除,加入新的培养液(加入培养基的量根据细胞的密度和生长速度);2、半量换液:把旧培养液移至15ml离心管中,然后在培养器皿中加入适量新培养基(避免细胞离开培养液太久),把装有旧培养液的离心管进行离心(1000RPM,10min),离心后取适量旧培养上清至培养器皿中。注意事项1.全量换液适合于生长较快的肿瘤细胞,因为这些细胞可在短期内分泌足够的因子来支撑其生长;2.半量换液主要适合于生长较慢的原代细胞和干细胞,这些细胞很难在短期内分泌足够的因子来支撑其生长,旧培养液中恰好含有这些因子;3.新旧培养液的配比取决于细胞的密度和生长速度及其自身的特性,请参照具体细胞的培养建议,一般为3:1(新:旧);4.如果细胞发生污染,切勿进行半量换液。三.细胞传代培养1、当细胞密度达到其生长密度极限时(一般肿瘤细胞为80-100%,原代细胞和干细胞为80-90%,有些细胞为40-50%,熟知所养细胞的生长密度极限),移除旧培养液;2、用PBS洗涤两次(旧培养中的钙离子会抑制胰酶的活性),加入1ml胰酶消化液(以T25培养瓶为例),立即盖好盖子。提供的原代细胞均需经过细胞类型特异性标记物、细胞形态学以及微生物检测等服务的公司。

血管生长是发生的关键步骤。无论原发性还是继发性,一旦生长直径超过1~2mm,都会有血管生成,这是由于细胞自身可分泌多种生长因子,诱导血管生成。由于组织这种新生血管结构及功能异常,且血管基质不完善,这种微血管容易发生渗漏,因此细胞不需经过复杂的侵袭过程而直接穿透到血管内进入血流并在远隔部位形成转移。越来越多的研究表明,良性血管生成稀少,血管生长缓慢;而大多数恶性的血管生成密集且生长迅速,因此,血管生成在的发展转移过程中起到重要作用,抑制这一过程将能明显阻止组织的发展和扩散转移。血管生成实验的技术原理主要是应用Matrigel模拟机体环境,上面接种细胞,观察血管生成情况。体外的血管生成实验能很好的模拟的血管发生过程,并且适合研究药物对这一过程的影响实验。我们以HUVEC细胞为例,介绍这一实验的详细过程。1、主要步骤Step1:细胞培养Step2:细胞转染或药物处理Step3:铺Matrigel胶Step4:接种细胞Step5:观察血管生成情况实验过程中需要注意:1、实验前一天需要将Matrigel置于冰盒,放入冰箱中,同时需要准备一些预冷的头用于吸取Matrigel胶;2、将Matrigel胶加到血管生成载玻片时注意头要垂直于内孔的正上方加入Matrigel。D大鼠食道平滑肌细胞分离自SD实验大鼠正常的食道组织,食道是消化管道的一部分。浙江纤维化原代细胞分离培养厂家
肝脏内的枯否细胞对于肝脏本身和全身的免疫应答都极为重要。云南C57小鼠原代细胞分离培养优势
把培养瓶置于倒置显微镜下观察,如大部分细胞变圆,立即移除胰酶消化液(如大部分细胞飘起,可不移除胰酶消化液),加入5ml预热完全培养基,用吸管取培养液吹打瓶壁细胞,使其脱离瓶壁形成单细胞悬液,离心,小心移除上清;3、加入适量(消化前预估细胞的数量,1-2*10E6/ml冻存液)冻存液并吹打混匀,分装至冻存管中,标记冻存细胞的名称、冷冻日期、操作者姓名拼音简写,然后放入梯度降温盒(提前复温至室温),置于-80℃过夜,次晨置于液氮罐中保存。注意事项1.密切观察细胞的生长密度,当细胞密度即将达到其生长密度极限时进行换液,以便第二天进行保种;2.消化前进行冻存液配制,切勿用培养基和血清重悬细胞后再加入DMSO,这样会导致局部高浓度的DMSO对细胞产生损伤;3.消化前预估细胞的数量,加入适量冻存液,以使细胞密度为1-2*10E6/ml,密度过高会导致冻存保护液不足,密度过低会导致冻存液对细胞有毒性;4.梯度降温盒必须提前复温至室温,切勿从-80℃取出即可使用,这样会导致细胞全部死亡;5.离心速度和时间依据具体细胞决定。环节问细胞体内外培养的差别是什么?答:细胞离体后,失去了神经体液的调节和细胞间的相互影响,生活在缺乏动态平衡相对稳定环境中。云南C57小鼠原代细胞分离培养优势
上一篇: 安徽口碑好的原代细胞分离培养哪家好
下一篇: 青海第三方原代细胞分离培养评价