黑龙江大鼠原代细胞分离培养评价
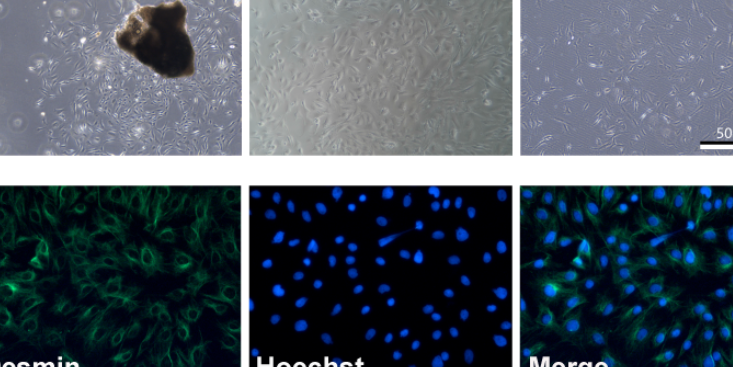
培养细胞不贴壁可能原因1.胰蛋白酶消化过度;2.支原体污染;3.培养基pH值过碱(NaHCO3分解);4.细胞老化;5.接种细胞起始浓度太低或太高。解决方法1.缩短胰蛋白酶消化时间或降低胰蛋白酶浓度;2.分离培养物,检测支原体。清洁支架或培养箱。如发现支原体污染,丢弃培养物;3.使用无菌醋酸溶液调整pH值或充入无菌CO2;4.启用新的保种细胞;5.调节接种细胞浓度。悬浮细胞成簇可能原因1.培养液中含钙,镁离子;2.支原体污染;3.蛋白酶过度消化使得细胞裂解;。解决方法1.用无钙镁平衡盐溶液洗涤细胞,轻巧吹吸2.细胞获得单细胞悬液;3.分离培养物,检测支原体;。培养细胞生长缓慢可能原因1.由于更换不同培养液或血清;2.培养液中一些细胞生长必须成分如谷氨酰胺或生长因子耗尽或缺乏或已被破坏;3.培养物中有少量细菌或污染;4.试剂保存不当;5.接种细胞起始浓度太低;6.细胞已老化;7.支原体污染。解决方法1.比较新培养液与原培养液成分,比较新血清与旧血清支持细胞生长实验,让细胞逐渐适应新培养液;2.换入新鲜配置培养液,或补加谷氨酰胺及生长因子;3.用无培养液培养,如发现污染,丢弃培养物;4.血清需保存在-10到-20℃。培养液需在2-8℃避光保存。肝脏星状细胞占肝脏固有细胞总数的15 %,占非实质细胞的30%左右。黑龙江大鼠原代细胞分离培养评价
如何判断是否加入了合适体积的Matrigel:我们只需用一张格子纸就能知道自己的移液调整到多少能正好把下孔填满。如上图所示,垂直透过每个孔看下面的格子纸,如果格子被缩小了,那么就说明胶没加满,格子被放大了,那么胶就加多了,格子没发生什么变形,这才是刚刚加满下孔的状态。2、凝胶1)盖上ibidi血管生成载玻片的盖子。2)准备一个10cm的培养皿,放入浸过水的纸巾,制成一个湿盒。3)将ibidi血管生成载玻片放入培养皿中,盖上培养皿盖。4)将整个培养皿放入培养箱中,静置30分钟左右,等待胶凝结。5)等待同时准备细胞悬液。3.铺细胞加入细胞的量直接影响实验结果,所以在正式实验开始之前,要对不同类型的细胞和使用数量进行预实验。获得比例的细胞密度。我们的实验使用HUVEC细胞,每孔种10000个细胞即可。1)准备密度为2×105的细胞悬液,充分混匀。2)将胶已经凝固的ibidi血管生成载玻片从湿盒中取出。3)每孔加入50μl的细胞悬液,注意保持头垂直在上孔的上方,不要接触下孔的凝胶。可以使用排。4)同样用格子纸查看是不是加了足够量的液体,如果没有,加入无细胞的培养基,使上孔液体正好加满。5)盖上盖,静置,一段时间后。宁夏怎样原代细胞分离培养评价肝实质细胞是指具有肝功能的基本组成单位,大约占肝脏体积及数量的80%左右。
一、原理在体外培养皿或平板培养的单层贴壁细胞上,用微量头或其它硬物在细胞生长的区域划线,去除部分的细胞,然后继续培养细胞至实验设定的时间,取出细胞培养板,在显微镜下观察并拍照记录特定位置细胞的生长迁移能力。通过不同分组之间的细胞对于划痕区修复能力的不同,可以判断各组细胞的迁移与修复能力的差别。二、步骤1、准备:所有能灭菌的器械都要灭菌,直尺和marker笔在操作前紫外照射30min(超净台内)2、流程:1.培养板划线:先用marker笔在6孔板背后,用直尺比着,均匀得划横线,大约每隔cm一道,横穿过孔,每孔至少穿过5条线;2.铺细胞:在孔中加入约5×105个细胞,具体数量因细胞不同而不同,掌握为过夜能铺满;3.细胞划线:第二天用头比着直尺,尽量垂至于背后的横线划痕,头要垂直,不能倾斜;4.洗细胞:用PBS洗细胞3次,去除划下的细胞,加入无血清培养基;5.细胞培养及观察:放入37℃、5%CO2培养箱,培养;按0,6,12,24小时取样,倒置显微镜观察特定位置细胞迁移情况并拍照;6.结果分析:使用ImageJ软件打开图片后,随机划取6-8条水平线,计算细胞间距离的均值。三、注意事项1、铺细胞使用6孔板,因为6孔板可以保证有相当距离的平直划痕。
1、取细胞县液100u加入Transwel小室。2、24孔板下室一般加入600u含20S的培养基,特别注意的是,下层培养液和小室间常会有气泡产生,一旦产生气泡,下层培养液的趋化作用就减弱其至消失了,在种板的时候要特别留心,一旦出现气泡,要将小室提起,去除气泡,再将小室放进培养板。3、培养细胞:常规培养12-48h(主要依细胞侵袭能力而定)。24h较常见,时间点的选择除了要考虑到细胞细胞侵袭力外,处理因素对细胞数目的影响也不可忽视。直接计数法贴壁”细胞计数,这里所谓的“贴“是指细胞穿过膜后,可以附着在膜的下室侧而不会掉到下室里面去,通讨给细胞染色可在镜下计数细胞。取出Transwel小室,弃去孔中培养液,用无钙的PBS洗2遍,用醇固定30分钟,将小室适当风干。01%结晶紫染色20min,用棉签轻轻掉上层未江移细胞,用PBS洗3遍。400倍显微镜下随即五人视野观察细胞,记数。内皮细胞在切应力的作用下,分泌不同的内皮因子并进而影响血管收缩和生长。
然后立即把细胞转移至提前准备的装有5-10ml预热的完全培养基的15ml离心管中;4、离心,小心移除上清;5、加入5ml预热完全培养基,吹打重悬细胞,然后把细胞悬液转移至T25培养瓶中,并放入二氧化碳培养箱(5%CO2,37℃)中培养;6、第二天观察细胞的贴壁情况,并进行换液。注意事项细胞复苏操作要求快速,否则细胞容易在冻融过程中产生冰晶,从而导致细胞死亡,所以必须提前准备好所需的培养基、培养耗材和其他所需物品,不允许临时准备;2.在解冻细胞前,必须把超净工作台准备至工作状态,提前准备装有5-10ml预热的完全培养基的15ml离心管;3.为了降低复温对其它细胞的影响,可把该存储架子放置于装有液氮的泡沫盒中;4.存储架离开液氮罐的时间一般不要超过1分钟,否则其余细胞容易复温死亡,冻存管子的液氮气化;5.放入之前快速检查盖子是否拧紧,盖子切勿没入水中,以免进水污染;6.细胞未冻融时(1min内)切勿取出观察;7.根据复苏细胞的大小选择合适的离心速度和时间,一般不超过1000RPM,10min;8.复苏后的第二天必须要进行换液(加入培养基的量根据细胞的密度和生长速度),以降低冻存保护剂DMSO的影响;9.离心速度和时间依据具体细胞决定;10.上述是以T25培养瓶为例。肾足细胞即肾小球上皮细胞分离技术。宁夏怎样原代细胞分离培养评价
D大鼠食道平滑肌细胞分离自SD实验大鼠正常的食道组织,食道是消化管道的一部分。黑龙江大鼠原代细胞分离培养评价
上期我们主要讲解了细胞凋亡的早期检测方法,这期主要讲解一下细胞凋亡晚期中,核酸内切酶(某些Caspase的底物)在核小体之间剪切核DNA,产生大量长度在180-200bp的DNA的片段。1、凋亡DNALadder检测试剂盒细胞经处理后,采用常规方法分离提纯DNA,进行琼脂糖凝胶和溴化乙啶染色,在凋亡细胞群中可观察到典型的DNAladder。QuickApoptoticDNALadderDetectionKit可以快速(约1-2小时)、灵敏地检测凋亡细胞中的DNA的片段。2、TUNEL-basedDNA的片段分析试剂盒细胞凋亡中,染色体DNA双链断裂或单链断裂而产生大量的粘性3-OH末端,可在脱氧核糖核苷酸末端转移酶(TdT)的作用下,将脱氧核糖核苷酸和荧光素、过氧化物酶、碱性磷酸酶或生物素形成的衍生物标记到DNA的3-末端,从而可进行凋亡细胞的检测。Apo-BrdUTM分析使用2种颜色的TUNEL方法用流式细胞仪检测凋亡细胞中DNA条带。采用的BrdU比生物素标记的或地高辛(digoxigenylated)标记的方法灵敏度更高。3、Caspase分析试剂和试剂盒Caspase在细胞凋亡中发挥着重要的作用,属于天冬氨酸蛋白酶。其中caspase-3为关键的执行分子,它在凋亡信号传导的许多途径中发挥功能。在正常状态下,caspase家族都以无活性的酶原。黑龙江大鼠原代细胞分离培养评价
上一篇: 海南面瘫原代细胞分离培养说明书
下一篇: 吉林哪里原代细胞分离培养供应商