杭州医疗器械CRO公司
医疗器械生产质量管理规范植入性医疗器械实施细则和检查评定标准,第五章设计和开发的具体要求(主要包括设计开发更改和文件记录)如下:第三十九条生产企业应当对设计和开发的更改进行识别并保持记录。必要时,应当对设计和开发更改进行评审、验证和确认,并在实施前得到批准。当选用的材料、零件或产品功能的改变可能影响到医疗器械产品安全性、有效性时,应当评价因改动可能带来的风险,必要时采取措施将风险降低到可接受水平,同时应当符合相关法规的要求。第四十条生产企业应当在包括设计和开发在内的产品实现全过程中,制定风险管理的要求并形成文件,保持相关记录。思脉得设计开发团队在满足相关法规要求的前提下,可根据客户需求进行定制化开发服务。医疗器械设计的成功离不开与专业技术服务商的密切合作。杭州医疗器械CRO公司
医疗器械设计数据管理平台内容分享:管理平台包括产品故障模式数据库。通过信息化管理,积累以往的经验教训,建立良好的设计数据管理平台,是实现研发标准化建设的基础,是提升产品研发质量管控能力的基础平台。好的设计成果和不合理的设计案例起到了示范作用。信息来源不仅包括研发过程的信息,更重要的是需要在后续的生产过程、安装、维护过程、客户使用过程中积累问题,逐步分析信息,总结造成产品质量差异的主要因素,提高设计开发质量和效率。医疗产品标志设计设计阶段的合规性考量是确保医疗器械上市的前提。

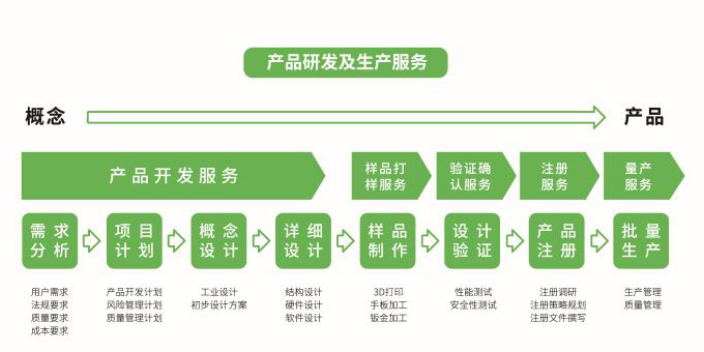
医疗器械设计开发是将医疗器械设计转化为商业可行产品的过程。在医疗器械行业,产品设计师必须在医疗器械设计的各个阶段,严格遵守法律法规要求,完整记录设计开发工作。在设计和开发策划的过程中,需要形成如下文件:1、设计和开发阶段;2、每个设计和开发阶段所需要的评审;3、适用于每个设计和开发阶段的验证、确认和设计转换活动;4、设计和开发的职责和权限;5、为确保设计和而开发输出到设计和开发输入可追溯性的方法;6、包括必要的人员能力在内的所需资源。从规划阶段开始,根据监管要求正式记录设计计划,描述开发过程,然后进入设计输入阶段。在这个阶段,设计过程中提出的要求和规格需要正式记录为用户需求和产品需求。
在ISO13485文件中明确了医疗器械设计开发的要求,主要从设计和开发策划、设计和开发输入、设计和开发输出、设计和开发评审、设计和开发验证、设计和开发确认、设计和开发转换、设计和开发更改的控制、设计和开发文件几方面进行了描述。思脉得医疗科技集团秉承创新技术和服务,为客户提供超预期价值为宗旨,致力于为医疗行业客户提供全套技术解决方案,为不同客户提供定制化的设计开发服务,涵盖产品需求分析、项目计划、概念设计、详细设计、样品制作、设计验证、产品注册等。如果您有相关需求,请与我们联系。CDMO服务商的全产业链支持可以帮助企业快速响应市场需求。
《医疗器械生产质量管理规范》中明确了设计开发过程的相关管理规范。下面按照各部分进行分享。设计开发策划:设计开发策划由整体项目规划、临床评价路径规划和风险管理规划几部分组成。目标、技术指标及其制定依据、设计开发阶段、各阶段的评审、验证和确认、职责权限和接口、风险管理。每个阶段的预期输出(文件和记录),每个阶段或任务所需的资源,以及完成每个阶段任务的预期时间范围。具体应包括:项目团队、人员职责、权限、任务、进度、从输入到输出的追溯方法;产品名称、预期用途/适用范围、分类、检测、临床、注册途径(是否申请创新或优先);人员职责、风险接受标准、各阶段风险管理活动、生产/生产后信息收集与审核。CDMO服务的兴起提升了整个医疗器械产业的服务水平。医疗产品设计
CDMO可以帮助企业更好地控制研发成本,提高资金利用效率。杭州医疗器械CRO公司
医疗器械生产质量管理规范植入性医疗器械实施细则和检查评定标准,第五章设计和开发的具体要求(主要包括设计开发评审和验证)如下:第三十六条生产企业应当在设计和开发的适宜阶段安排评审,保持评审结果及任何必要措施的记录。第三十七条生产企业应当对设计和开发进行验证,以确保设计和开发输出满足输入的要求,并保持验证结果和任何必要措施的记录。第三十八条生产企业应当对设计和开发进行确认,以确保产品满足规定的使用要求或预期用途的要求,并保持确认结果和任何必要措施的记录。确认可采用临床评价和/或性能评价。进行临床试验时应当符合医疗器械临床试验法规的要求。杭州医疗器械CRO公司
上一篇: 安徽医疗器械设计开发常见问题
下一篇: 苏州医疗器械设计开发常见问题